
Understanding Protein Language Models Part I: Multiple Sequence Alignment in AlphaFold2
The fundamental concepts behind ESM2, ESM3, and AlphaFold


Note: This post is part of the “Understanding Protein Language Models” Series:
[This article] Understanding Protein Language Models Part I: Multiple Sequence Alignment in AlphaFold2
Over the past couple of months, I’ve been on a journey to understand protein language models - what exactly are they learning and how do they work? I began that journey by trying to understand the inner working of AlphaFold2, given that it represented the first leap forward for AI in biology.
In particular, protein language models sought to replace the multiple sequence alignment (MSA) component of AlphaFold2 in order to achieve similar performance at much lower computational costs. So, in order to better understand what protein language models are doing, it is first important to understand what they replaced. To that end, in these notes I dive into the role of multiple sequence alignments in AlphaFold2 and how it drives the model’s ability to infer contacts between residues in the protein. I hope you find these notes useful!
What is MSA?

Multiple sequence alignment (MSA) refers to the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a linkage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins.
Visual depictions of the alignment (as in the image below) illustrate mutation events such as point mutations (single amino acid or nucleotide changes) that appear as differing characters in a single alignment column, and insertion or deletion mutations (indels or gaps) that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.
Thus, by including an MSA as input, AlphaFold2 is able to infer information about the target sequence by assessing its shared evolutionary history with a number of other sequences. This is a powerful concept - it gives the model a strong “starting point” to make predictions for a new sequence.
The MSA databases used by AlphaFold2 to identify evolutionarily similar sequences to the target sequence are:
- MGnify
- UniRef90
- Uniclust30
- BFD
AlphaFold2 Architecture

Now that we know what MSA is and why it is used, let’s sketch out the high-level architectural details of AlphaFold2. In the above image, we can see the various inputs & components that comprise the model. AlphaFold2 takes in three key inputs:
The input sequence itself
An MSA using the input sequence as its starting point
Template structure related to the input sequence
These three inputs are then distilled into two by using the template structures and input sequence to initialize a pair representation matrix. The pair representation matrix can be thought of as scores for “similarity” or “interaction” between each pair of amino acids i and j in the input sequence.
By contrast, the MSA representation can be thought of as storing a vector representation of each amino acid for each protein in the alignment. If we imagine the matrix as a 2D grid, each row represents a protein and column represents a position in the aligned amino acid sequence (e.g. amino acid #5 in the sequence). In each cell of this matrix, we can imagine a vector that represents the specified amino acid. In reality, this is a tensor of shape (number of sequences, number of residues, channels).
These two inputs then flow through the Evoformer block, which generates improved representations of the MSA and pair representation matrices for structure prediction. The journey for the full MSA matrix ends here, as we extract the representation for our input sequence from the first row of the MSA matrix and send it forward to the structure module.
Given that the processing for the MSA matrix takes place in the Evoformer block, we’ll dive in a bit deeper there.
Evoformer Block

The Evoformer block begins with components for processing the MSA representation:
Row-wise gated self-attention
Column-wise gated self-attention
Transition
Following these three blocks, the MSA representation matrix is integrated into the pair representation matrix through the outer product mean block and resulting sum.
We’ll now dive deep into the three core components of the Evoformer block (alongisde the outer product mean integration) to better understand how the MSA matrix is being updated.
MSA Row-wise Gated Self-Attention
Row-wise attention builds attentions weights for residue pairs within the same sequence and integrates the information from the pair representation as an additional bias term. The updated MSA representation matrix thus ensures that each sequence has a contextual representation for its residues - that is, for sequence k, the embedding of the residue at index i takes into account information from the residues at indices 1, …, i-1, i+1, …, r

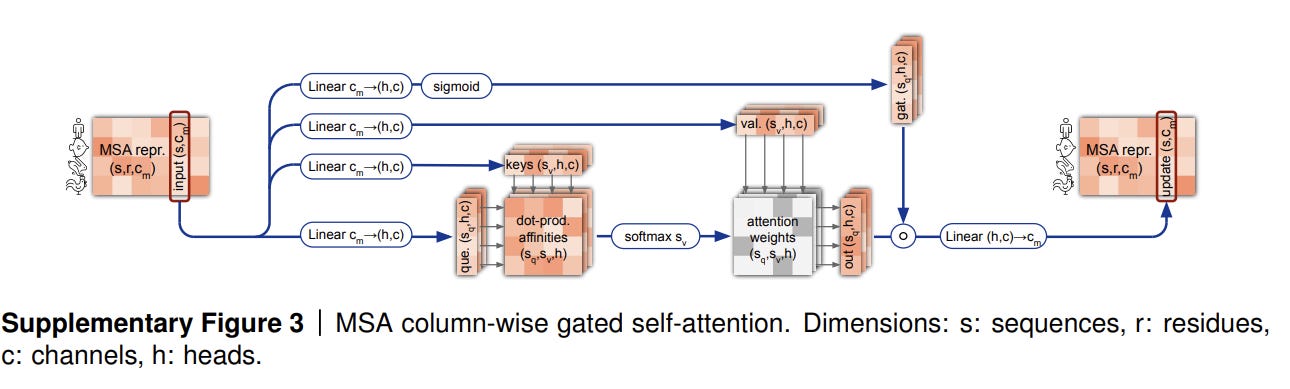
MSA Column-wise Gated Self-Attention
Column-wise attention lets the elements that belong to the same target residue exchange information across sequences in the MSA. The updated MSA representation matrix thus ensures that each residue has a cross-sequence representation - that is, for the embedding for residue i in sequence k also takes into account information from residue i in sequences 1, …, k-1, k+1, …, s.
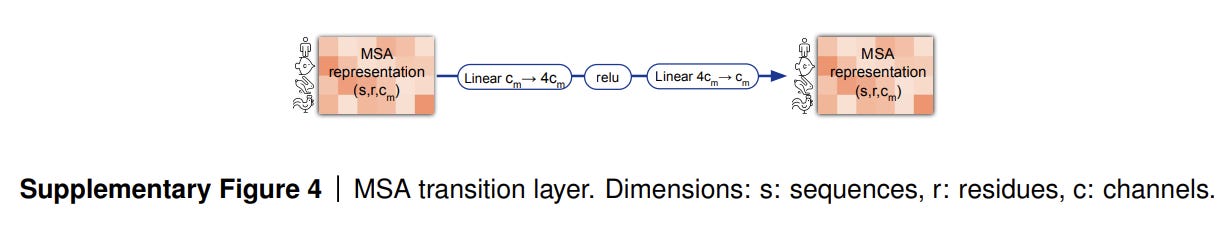
MSA Transition
After row-wise and column-wise attention the MSA stack contains a 2-layer MLP as the transition layer. The intermediate number of channels expands the original number of channels by a factor of 4.
Integration of MSA with Pair Representation
The “Outer product mean” block transforms the MSA representation into an update for the pair representation.

In particular, this step grabs two vectors of representations for residues i and j, where the vectors span the representations of all sequences included in the MSA. The outer product step creates a matrix of all dot product combinations. In the below image, you can think of u_1 as "sequence 1, residue i", u_2 as "sequence 2, residue i", and so on. Similarly, you can think of v_1 as "sequence 1, residue j", v_2 as "sequence 2, residue j", and so on. Since these dot products gives us a measure of the similarity between the representation of "sequence k, residue i" and "sequence m, residue j", we can think of it as a matrix that captures the pairwise similarities between all residues at positions i and j in the MSA.

This matrix ends up being of shape (s, c, c), where s is the number of sequences and each c dimension denotes the number of features for residue i and j's representations, respectively. AlphaFold then takes a mean over the s dimension of the matrix. What this means intuitively is that we average the pairwise similarity of residue i and residue j across all possible pairs of sequences in the MSA matrix. This collapses matrix from shape (s, c, c) to shape (c, c).
The last step projects the features to from (c, c) to c_z. This allows them to be added to each entry in the pairwise representation.
Conclusion - What is the MSA Representation Doing in AlphaFold?
So, putting this all together - the MSA steps compute a representation that optimally captures similarity of residues, both:
1. Within sequences by using row-wise attention to attend across amino acids inside a given sequence
2. Across sequences by using column-wise attention to attend across sequences for a given amino acid index
This representation is then used to generate a measure of similarity between all possible residue pairs in the MSA representation. We then update the pair representation of the target sequence by adding in these values. In essence, we use the MSA to "find out" which residues are similar to which other residues, and then add this information to the pair representation so that the structure module can guess at which residues are in contact with one another (based on the fact that they co-evolve and are therefore similar in the MSA representation). This allows for highly accurate structure prediction, incorporating information from the evolutionary tree to infer the optimal folded structure of a given input protein.